Retrieves "semantic phenotypes", i.e., phenotypes encoded as ontological
expressions. Filtering is possible by anatomical entity (optionally including
entities related by certain properties, see includeRels), phenotypic
quality, taxonomic group where the phenotypes have been recorded, and study
(a.k.a. publication).
get_phenotypes(
entity = NA,
quality = NA,
taxon = NA,
study = NA,
includeRels = c("part of"),
.withTaxon = FALSE,
verbose = FALSE
)Arguments
- entity
character, the anatomical entity by which to filter, if any.
- quality
character, the phenotypic quality by which to filter, if any.
- taxon
character, the taxon by which to filter, if any.
- study
character, the identifier of the study by which to filter, if any.
- includeRels
character or vector of characters. The names of relationships for anatomical entities to include in addition to subtype (
rdfs:subClassOf). Defaults to"part of". Set toFALSEto not include any additional relationships. Otherwise one or more of"part of","historical homologous to", and"serially homologous to", or set toTRUEto include all possible ones. It is acceptable to use unambiguous prefixes, for example"historical".- .withTaxon
logical, whether to include taxa in the result if
taxonis provided. If TRUE, only the combination of phenotype and taxon will be unique in the returned data frame. Default is FALSE, meaning by default providing a value fortaxononly acts as another filter but does not change format or redundancy of the result. Ignored iftaxonis not provided as a character value.- verbose
logical, whether to print messages informing about potentially time-consuming operations. Default is FALSE.
Value
A data frame with columns "id" and "label".
If a character value for taxon was provided, and .withTaxon is TRUE’,
columns "taxon.id" and "taxon.label" will be returned as well. While
(phenotypes, taxon) tuples will be unique, both phenotypes and taxa
individually will then be redundant in the returned data frame (the
association is n:n).
Details
Entity, quality, and taxon can be given as IRI or as name (i.e, term label). In the latter case, names will be resolved to IRIs against anatomy ontologies, PATO, and taxonomy ontologies, respectively. Warnings will be issued if only a partial match can be found. The study must be given as IRI.
Examples
phens1 <- get_phenotypes(entity = "pelvic fin")
head(phens1)
#> id
#> 1 http://purl.org/phenoscape/expression?value=%3Chttp%3A%2F%2Fpurl.obolibrary.org%2Fobo%2FBFO_0000051%3E+some+%0A++++%28%3Chttp%3A%2F%2Fpurl.obolibrary.org%2Fobo%2FPATO_0000140%3E%0A+++++and+%28%3Chttp%3A%2F%2Fpurl.obolibrary.org%2Fobo%2FRO_0000052%3E+some+%3Chttp%3A%2F%2Fpurl.obolibrary.org%2Fobo%2FUBERON_2001254%3E%29%0A+++++and+%28%3Chttp%3A%2F%2Fpurl.obolibrary.org%2Fobo%2FRO_0002503%3E+some+%3Chttp%3A%2F%2Fpurl.obolibrary.org%2Fobo%2FUBERON_0000152%3E%29%29
#> 2 http://purl.org/phenoscape/expression?value=%28%3Chttp%3A%2F%2Fpurl.obolibrary.org%2Fobo%2FBFO_0000051%3E+some+%0A++++%28%3Chttp%3A%2F%2Fpurl.obolibrary.org%2Fobo%2FPATO_0002000%3E%0A+++++and+%28%3Chttp%3A%2F%2Fpurl.obolibrary.org%2Fobo%2FRO_0000052%3E+some+%3Chttp%3A%2F%2Fpurl.obolibrary.org%2Fobo%2FUBERON_0000468%3E%29%0A+++++and+%28%3Chttp%3A%2F%2Fpurl.obolibrary.org%2Fobo%2FRO_0002503%3E+value+%3Chttp%3A%2F%2Fpurl.org%2Fphenoscape%2Fsubclassof%3Fvalue%3D%253Chttp%253A%252F%252Fpurl.obolibrary.org%252Fobo%252FUBERON_0004529%253E%250A%2Band%2B%2528%253Chttp%253A%252F%252Fpurl.obolibrary.org%252Fobo%252FBFO_0000050%253E%2Bsome%2B%250A%2B%2B%2B%2B%2528%253Chttp%253A%252F%252Fpurl.obolibrary.org%252Fobo%252FBSPO_0000029%253E%250A%2B%2B%2B%2B%2Band%2B%2528%253Chttp%253A%252F%252Fpurl.obolibrary.org%252Fobo%252FBFO_0000050%253E%2Bsome%2B%250A%2B%2B%2B%2B%2B%2B%2B%2B%2528%253Chttp%253A%252F%252Fpurl.obolibrary.org%252Fobo%252FBSPO_0000383%253E%250A%2B%2B%2B%2B%2B%2B%2B%2B%2Band%2B%2528%253Chttp%253A%252F%252Fpurl.obolibrary.org%252Fobo%252FBFO_0000050%253E%2Bsome%2B%253Chttp%253A%252F%252Fpurl.obolibrary.org%252Fobo%252FUBERON_4000173%253E%2529%2529%2529%2529%2529%23aba77fda-e5de-4fd0-b369-ec542b79429b%3E%29%29%29%0A+and+%28%3Chttp%3A%2F%2Fpurl.org%2Fphenoscape%2Fvocab.owl%23phenotype_of%3E+some+%3Chttp%3A%2F%2Fpurl.org%2Fphenoscape%2Fsubclassof%3Fvalue%3D%253Chttp%253A%252F%252Fpurl.obolibrary.org%252Fobo%252FUBERON_0004529%253E%250A%2Band%2B%2528%253Chttp%253A%252F%252Fpurl.obolibrary.org%252Fobo%252FBFO_0000050%253E%2Bsome%2B%250A%2B%2B%2B%2B%2528%253Chttp%253A%252F%252Fpurl.obolibrary.org%252Fobo%252FBSPO_0000029%253E%250A%2B%2B%2B%2B%2Band%2B%2528%253Chttp%253A%252F%252Fpurl.obolibrary.org%252Fobo%252FBFO_0000050%253E%2Bsome%2B%250A%2B%2B%2B%2B%2B%2B%2B%2B%2528%253Chttp%253A%252F%252Fpurl.obolibrary.org%252Fobo%252FBSPO_0000383%253E%250A%2B%2B%2B%2B%2B%2B%2B%2B%2Band%2B%2528%253Chttp%253A%252F%252Fpurl.obolibrary.org%252Fobo%252FBFO_0000050%253E%2Bsome%2B%253Chttp%253A%252F%252Fpurl.obolibrary.org%252Fobo%252FUBERON_4000173%253E%2529%2529%2529%2529%2529%23aba77fda-e5de-4fd0-b369-ec542b79429b%3E%29
#> 3 http://purl.org/phenoscape/expression?value=%3Chttp%3A%2F%2Fpurl.obolibrary.org%2Fobo%2FBFO_0000051%3E+some+%0A++++%28%3Chttp%3A%2F%2Fpurl.obolibrary.org%2Fobo%2FPATO_0000600%3E%0A+++++and+%28%3Chttp%3A%2F%2Fpurl.obolibrary.org%2Fobo%2FRO_0000052%3E+some+%3Chttp%3A%2F%2Fpurl.org%2Fphenoscape%2Fsubclassof%3Fvalue%3D%253Chttp%253A%252F%252Fpurl.obolibrary.org%252Fobo%252FUBERON_0004529%253E%250A%2Band%2B%2528%253Chttp%253A%252F%252Fpurl.obolibrary.org%252Fobo%252FBFO_0000050%253E%2Bsome%2B%250A%2B%2B%2B%2B%2528%253Chttp%253A%252F%252Fpurl.obolibrary.org%252Fobo%252FBSPO_0000029%253E%250A%2B%2B%2B%2B%2Band%2B%2528%253Chttp%253A%252F%252Fpurl.obolibrary.org%252Fobo%252FBFO_0000050%253E%2Bsome%2B%250A%2B%2B%2B%2B%2B%2B%2B%2B%2528%253Chttp%253A%252F%252Fpurl.obolibrary.org%252Fobo%252FBSPO_0000383%253E%250A%2B%2B%2B%2B%2B%2B%2B%2B%2Band%2B%2528%253Chttp%253A%252F%252Fpurl.obolibrary.org%252Fobo%252FBFO_0000050%253E%2Bsome%2B%253Chttp%253A%252F%252Fpurl.obolibrary.org%252Fobo%252FUBERON_4000173%253E%2529%2529%2529%2529%2529%231c9c6eec-ba1b-4ee6-8f8d-f95fde562e67%3E%29%29
#> 4 http://purl.org/phenoscape/expression?value=%3Chttp%3A%2F%2Fpurl.obolibrary.org%2Fobo%2FBFO_0000051%3E+some+%0A++++%28%3Chttp%3A%2F%2Fpurl.obolibrary.org%2Fobo%2FPATO_0000052%3E%0A+++++and+%28%3Chttp%3A%2F%2Fpurl.obolibrary.org%2Fobo%2FRO_0000052%3E+some+%3Chttp%3A%2F%2Fpurl.org%2Fphenoscape%2Fsubclassof%3Fvalue%3D%253Chttp%253A%252F%252Fpurl.obolibrary.org%252Fobo%252FUBERON_0004529%253E%250A%2Band%2B%2528%253Chttp%253A%252F%252Fpurl.obolibrary.org%252Fobo%252FBFO_0000050%253E%2Bsome%2B%250A%2B%2B%2B%2B%2528%253Chttp%253A%252F%252Fpurl.obolibrary.org%252Fobo%252FBSPO_0000029%253E%250A%2B%2B%2B%2B%2Band%2B%2528%253Chttp%253A%252F%252Fpurl.obolibrary.org%252Fobo%252FBFO_0000050%253E%2Bsome%2B%250A%2B%2B%2B%2B%2B%2B%2B%2B%2528%253Chttp%253A%252F%252Fpurl.obolibrary.org%252Fobo%252FBSPO_0000383%253E%250A%2B%2B%2B%2B%2B%2B%2B%2B%2Band%2B%2528%253Chttp%253A%252F%252Fpurl.obolibrary.org%252Fobo%252FBFO_0000050%253E%2Bsome%2B%253Chttp%253A%252F%252Fpurl.obolibrary.org%252Fobo%252FUBERON_4000173%253E%2529%2529%2529%2529%2529%232f682357-b062-4b46-b5fd-2027f1709ae5%3E%29%29
#> 5 http://purl.org/phenoscape/expression?value=%28%3Chttp%3A%2F%2Fpurl.obolibrary.org%2Fobo%2FBFO_0000051%3E+some+%0A++++%28%3Chttp%3A%2F%2Fpurl.obolibrary.org%2Fobo%2FPATO_0002000%3E%0A+++++and+%28%3Chttp%3A%2F%2Fpurl.obolibrary.org%2Fobo%2FRO_0000052%3E+some+%3Chttp%3A%2F%2Fpurl.obolibrary.org%2Fobo%2FUBERON_0000468%3E%29%0A+++++and+%28%3Chttp%3A%2F%2Fpurl.obolibrary.org%2Fobo%2FRO_0002503%3E+value+%3Chttp%3A%2F%2Fpurl.org%2Fphenoscape%2Fsubclassof%3Fvalue%3D%253Chttp%253A%252F%252Fpurl.obolibrary.org%252Fobo%252FUBERON_0004529%253E%250A%2Band%2B%2528%253Chttp%253A%252F%252Fpurl.obolibrary.org%252Fobo%252FBFO_0000050%253E%2Bsome%2B%250A%2B%2B%2B%2B%2528%253Chttp%253A%252F%252Fpurl.obolibrary.org%252Fobo%252FBSPO_0000079%253E%250A%2B%2B%2B%2B%2Band%2B%2528%253Chttp%253A%252F%252Fpurl.obolibrary.org%252Fobo%252FBFO_0000050%253E%2Bsome%2B%253Chttp%253A%252F%252Fpurl.obolibrary.org%252Fobo%252FUBERON_0000152%253E%2529%2529%2529%235eeaadfb-dc08-4924-b223-df64eae0b1d2%3E%29%29%29%0A+and+%28%3Chttp%3A%2F%2Fpurl.org%2Fphenoscape%2Fvocab.owl%23phenotype_of%3E+some+%3Chttp%3A%2F%2Fpurl.org%2Fphenoscape%2Fsubclassof%3Fvalue%3D%253Chttp%253A%252F%252Fpurl.obolibrary.org%252Fobo%252FUBERON_0004529%253E%250A%2Band%2B%2528%253Chttp%253A%252F%252Fpurl.obolibrary.org%252Fobo%252FBFO_0000050%253E%2Bsome%2B%250A%2B%2B%2B%2B%2528%253Chttp%253A%252F%252Fpurl.obolibrary.org%252Fobo%252FBSPO_0000079%253E%250A%2B%2B%2B%2B%2Band%2B%2528%253Chttp%253A%252F%252Fpurl.obolibrary.org%252Fobo%252FBFO_0000050%253E%2Bsome%2B%253Chttp%253A%252F%252Fpurl.obolibrary.org%252Fobo%252FUBERON_0000152%253E%2529%2529%2529%235eeaadfb-dc08-4924-b223-df64eae0b1d2%3E%29
#> 6 http://purl.org/phenoscape/expression?value=%3Chttp%3A%2F%2Fpurl.obolibrary.org%2Fobo%2FBFO_0000051%3E+some+%0A++++%28%3Chttp%3A%2F%2Fpurl.obolibrary.org%2Fobo%2FPATO_0000569%3E%0A+++++and+%28%3Chttp%3A%2F%2Fpurl.obolibrary.org%2Fobo%2FRO_0000052%3E+some+%3Chttp%3A%2F%2Fpurl.org%2Fphenoscape%2Fsubclassof%3Fvalue%3D%253Chttp%253A%252F%252Fpurl.obolibrary.org%252Fobo%252FUBERON_0004529%253E%250A%2Band%2B%2528%253Chttp%253A%252F%252Fpurl.obolibrary.org%252Fobo%252FBFO_0000050%253E%2Bsome%2B%250A%2B%2B%2B%2B%2528%253Chttp%253A%252F%252Fpurl.obolibrary.org%252Fobo%252FBSPO_0000079%253E%250A%2B%2B%2B%2B%2Band%2B%2528%253Chttp%253A%252F%252Fpurl.obolibrary.org%252Fobo%252FBFO_0000050%253E%2Bsome%2B%253Chttp%253A%252F%252Fpurl.obolibrary.org%252Fobo%252FUBERON_0000152%253E%2529%2529%2529%233c7a478b-5bb2-4378-a88f-10e007f020b1%3E%29%29
#> label
#> 1 abdominal scute series position pelvic fin
#> 2 anatomical projection and (part_of some (antero-lateral region and (part_of some (medial surface and (part_of some pelvic fin lepidotrichium))))) absent
#> 3 anatomical projection and (part_of some (antero-lateral region and (part_of some (medial surface and (part_of some pelvic fin lepidotrichium))))) increased width
#> 4 anatomical projection and (part_of some (antero-lateral region and (part_of some (medial surface and (part_of some pelvic fin lepidotrichium))))) shape
#> 5 anatomical projection and (part_of some (dorsal region and (part_of some pelvic fin))) absent
#> 6 anatomical projection and (part_of some (dorsal region and (part_of some pelvic fin))) decreased height
# by default, parts are already included
phens2 <- get_phenotypes(entity = "pelvic fin", includeRels = c("part"))
nrow(phens1) == nrow(phens2)
#> [1] TRUE
table(phens2$id %in% phens1$id)
#>
#> TRUE
#> 227
# but historical homologues are not
phens2 <- get_phenotypes(entity = "pelvic fin", includeRels = c("part", "hist"))
table(phens2$id %in% phens1$id)
#>
#> FALSE TRUE
#> 1173 227
# neither are serially homologous
phens2 <- get_phenotypes(entity = "pelvic fin", includeRels = TRUE)
table(phens2$id %in% phens1$id)
#>
#> FALSE TRUE
#> 1622 227
# filter also by quality
phens2 <- get_phenotypes(entity = "pelvic fin", quality = "shape")
table(phens1$id %in% phens2$id)
#>
#> FALSE TRUE
#> 199 28
# filter also by quality and taxon
phens2 <- get_phenotypes(entity = "pelvic fin", quality = "shape", taxon = "Siluriformes")
table(phens1$id %in% phens2$id)
#>
#> FALSE TRUE
#> 215 12
# filter by entity, quality and taxon, and return taxa as well (resulting in
# (phenotype, taxon) "tuples")
phens2a <- get_phenotypes(entity = "pelvic fin", quality = "shape", taxon = "Siluriformes",
.withTaxon = TRUE)
head(phens2a)
#> id
#> 1 http://purl.org/phenoscape/expression?value=%3Chttp%3A%2F%2Fpurl.obolibrary.org%2Fobo%2FBFO_0000051%3E+some+%0A++++%28%3Chttp%3A%2F%2Fpurl.obolibrary.org%2Fobo%2FPATO_0000407%3E%0A+++++and+%28%3Chttp%3A%2F%2Fpurl.obolibrary.org%2Fobo%2FRO_0000052%3E+some+%3Chttp%3A%2F%2Fpurl.obolibrary.org%2Fobo%2FUBERON_2001776%3E%29%29
#> 2 http://purl.org/phenoscape/expression?value=%3Chttp%3A%2F%2Fpurl.obolibrary.org%2Fobo%2FBFO_0000051%3E+some+%0A++++%28%3Chttp%3A%2F%2Fpurl.obolibrary.org%2Fobo%2FPATO_0000414%3E%0A+++++and+%28%3Chttp%3A%2F%2Fpurl.obolibrary.org%2Fobo%2FRO_0000052%3E+some+%3Chttp%3A%2F%2Fpurl.obolibrary.org%2Fobo%2FUBERON_2001781%3E%29%29
#> 3 http://purl.org/phenoscape/expression?value=%3Chttp%3A%2F%2Fpurl.obolibrary.org%2Fobo%2FBFO_0000051%3E+some+%0A++++%28%3Chttp%3A%2F%2Fpurl.obolibrary.org%2Fobo%2FPATO_0000414%3E%0A+++++and+%28%3Chttp%3A%2F%2Fpurl.obolibrary.org%2Fobo%2FRO_0000052%3E+some+%3Chttp%3A%2F%2Fpurl.obolibrary.org%2Fobo%2FUBERON_2001781%3E%29%29
#> 4 http://purl.org/phenoscape/expression?value=%3Chttp%3A%2F%2Fpurl.obolibrary.org%2Fobo%2FBFO_0000051%3E+some+%0A++++%28%3Chttp%3A%2F%2Fpurl.obolibrary.org%2Fobo%2FPATO_0000052%3E%0A+++++and+%28%3Chttp%3A%2F%2Fpurl.obolibrary.org%2Fobo%2FRO_0000052%3E+some+%3Chttp%3A%2F%2Fpurl.obolibrary.org%2Fobo%2FUBERON_2002077%3E%29%29
#> 5 http://purl.org/phenoscape/expression?value=%3Chttp%3A%2F%2Fpurl.obolibrary.org%2Fobo%2FBFO_0000051%3E+some+%0A++++%28%3Chttp%3A%2F%2Fpurl.obolibrary.org%2Fobo%2FPATO_0000052%3E%0A+++++and+%28%3Chttp%3A%2F%2Fpurl.obolibrary.org%2Fobo%2FRO_0000052%3E+some+%3Chttp%3A%2F%2Fpurl.obolibrary.org%2Fobo%2FUBERON_2001776%3E%29%29
#> 6 http://purl.org/phenoscape/expression?value=%3Chttp%3A%2F%2Fpurl.obolibrary.org%2Fobo%2FBFO_0000051%3E+some+%0A++++%28%3Chttp%3A%2F%2Fpurl.obolibrary.org%2Fobo%2FPATO_0000052%3E%0A+++++and+%28%3Chttp%3A%2F%2Fpurl.obolibrary.org%2Fobo%2FRO_0000052%3E+some+%3Chttp%3A%2F%2Fpurl.obolibrary.org%2Fobo%2FUBERON_2002077%3E%29%29
#> label taxon.id
#> 1 pelvic fin ray 1 flat http://purl.obolibrary.org/obo/VTO_0035629
#> 2 pelvic fin ray 6 unbranched http://purl.obolibrary.org/obo/VTO_0035629
#> 3 pelvic fin ray 6 unbranched http://purl.obolibrary.org/obo/VTO_0060892
#> 4 lateropterygium shape http://purl.obolibrary.org/obo/VTO_0035553
#> 5 pelvic fin ray 1 shape http://purl.obolibrary.org/obo/VTO_0035553
#> 6 lateropterygium shape http://purl.obolibrary.org/obo/VTO_0061318
#> taxon.label
#> 1 Acentronichthys leptos
#> 2 Acentronichthys leptos
#> 3 Acentronichthys sp. n. B (Bockmann 1998)
#> 4 Acrochordonichthys rugosus
#> 5 Acrochordonichthys rugosus
#> 6 Acrochordonichthys sp. (de Pinna 1993)
nrow(phens2a) - nrow(phens2) # lots of redundancy due to n:n relationship
#> [1] 963
nrow(unique(phens2a[,c("id", "label")])) == nrow(phens2) # but same #phenotypes
#> [1] TRUE
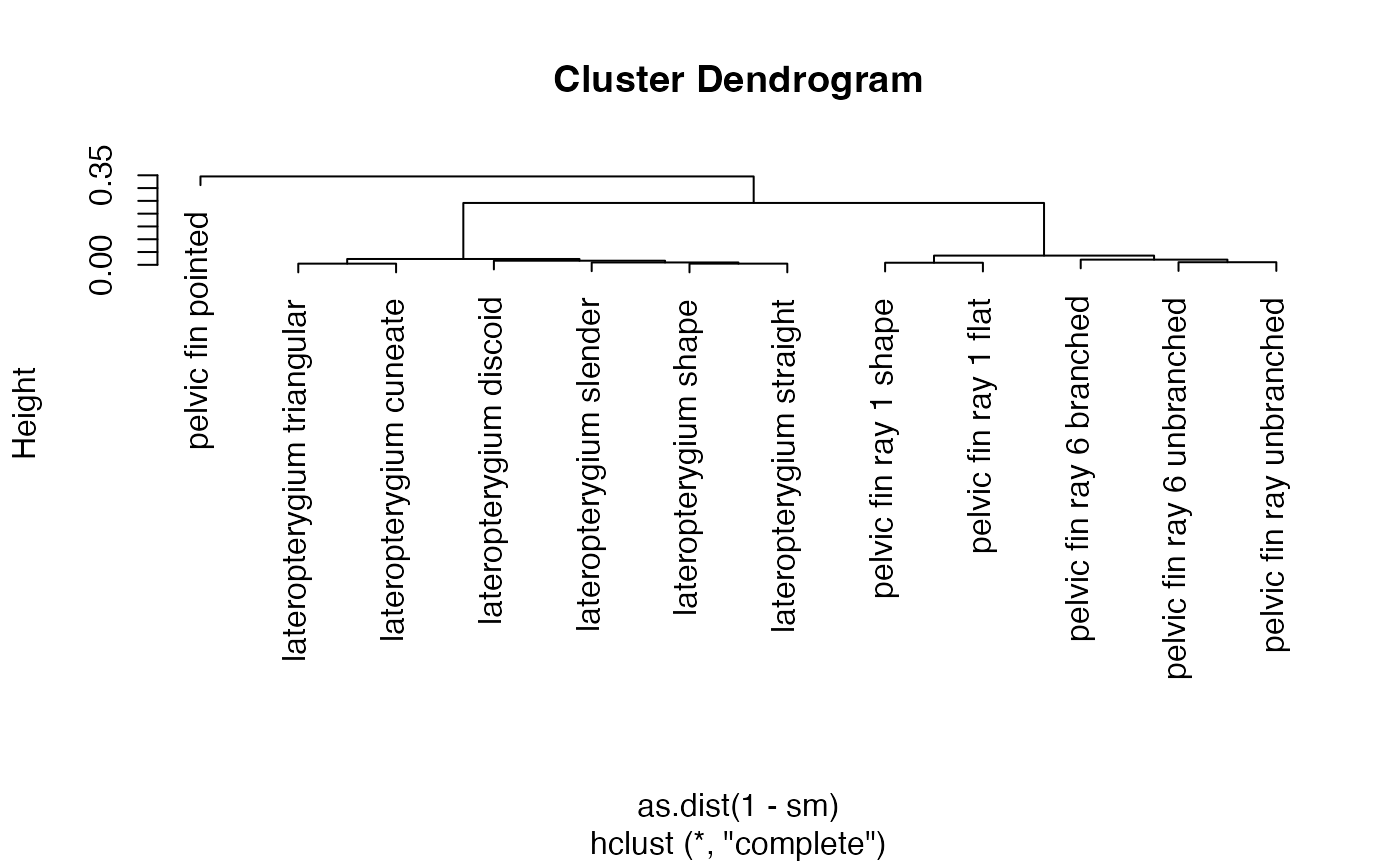
# can compute and visualize similarity
sm <- jaccard_similarity(terms = phens2$id, .labels = phens2$label, .colnames = "label")
plot(hclust(as.dist(1-sm)))